Centre régional de dépistage néonatal d'Île-de-France (CRDN-IdF) & Coordination francilienne du dépistage néonatal de l’audition (CFDNA)

![]() Le programme national de dépistage néonatal (DNN) concerne tous les nouveau-nés qui naissent en France. Il vise à détecter sur un échantillon de sang total recueilli sur buvard (Guthrie) à 3 jours de vie et à l’aide des examens de biologie médicale (défini par l’arrêté du 9 novembre 2022 modifiant l’arrêté du 12 novembre 2020 modifiant l’arrêté du 22 février 2018, relatif à l’organisation du Programme National de Dépistage Néonatal recourant à des examens de biologie médicale) ) des maladies rares, sévères et le plus souvent génétiques.
Le programme national de dépistage néonatal (DNN) concerne tous les nouveau-nés qui naissent en France. Il vise à détecter sur un échantillon de sang total recueilli sur buvard (Guthrie) à 3 jours de vie et à l’aide des examens de biologie médicale (défini par l’arrêté du 9 novembre 2022 modifiant l’arrêté du 12 novembre 2020 modifiant l’arrêté du 22 février 2018, relatif à l’organisation du Programme National de Dépistage Néonatal recourant à des examens de biologie médicale) ) des maladies rares, sévères et le plus souvent génétiques.
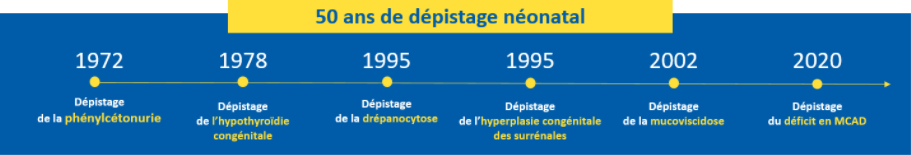
Le but du DNN est de prévenir la survenue de manifestations et de complications graves dues aux maladies dépistées afin, a minima, d’en limiter la gravité. Parce qu’il existe des moyens thérapeutiques efficaces, le dépistage précoce apporte un véritable bénéfice aux nouveau-nés. Dans un délai bref, il est possible de confirmer le diagnostic définitif et de mettre en place, très tôt après la naissance, le traitement adéquat. Par exemple, le diagnostic de la phénylcétonurie (PCU) à la naissance permet grâce à un régime et à un suivi médical adéquats, d’éviter la survenue d’un handicap cognitif sévère et définitif.
En 2021, le programme de DNN a permis de dépister en France :
- 284 hypothyroïdies congénitales dont 52 (soit 18%) en IdF;
- 35 hyperplasies congénitales des surrénales dont 9 (soit 26%) en IdF ;
- 623 syndromes drépanocytaires majeurs dont 314 (soit 50) en IdF ;
- 118 mucoviscidoses dont 17 (soit 14%) en IdF ;
- 46 phénylcétonuries et 66 hyperphénylalaninémies modérées permanentes, dont 10 (soit 22%) PCU et 20 HPMP (soit 30%) en IdF ;
- Depuis le 1er décembre 2020, 28 déficits en acyl-CoA déshydrogénase des acides gras à chaîne moyenne (MCAD) dont 8 (soit 28%) en IdF.
L’introduction des techniques de spectrométrie de masse en tandem a permis l’extension du dépistage à d’autres erreurs innées du métabolisme. Au 1er janvier 2023, sont inclues les maladies suivantes :
- 3 autres aminoacidopathies : homocystinurie classique (HCY), leucinose (MSUD) et tyrosinémie de type 1 (HT-1) ;
- 2 autres déficits de la bêta-oxydation : déficit en déshydrogénase des hydroxyacyl-CoA des acides gras à chaine longue (LCHAD) et déficit en captation de la carnitine libre (CUD) ;
- 2 aciduries organiques : acidurie glutarique de type 1 (AG-1) et acidurie isovalérique (IVA).
Le programme biologique est complété par le dépistage de la surdité permanente néonatale, qui n’est pas organisé par le CRDN-IdF mais par les réseaux de périnatalité coordonnés par la coordination francilienne de dépistage néonatal de l’audition (CFDNA) en lien avec l’ARS.
> Tableau des caractéristiques des 13 maladies dépistées en France
> Consulter le site du programme national de dépistage néonatal
Mots clés : dépistage néonatal, prévention, maladies rares, phénylcétonurie, hypothyroïdie congénitale, hyperplasie congénitale des surrénales, drépanocytose, mucoviscidose, maladies héréditaires du métabolisme.
Médecin
responsable

Pr Michel Polak
Contact
.
Hôpital universitaire Necker-Enfants malades
Centre régional de dépistage néonatal
149 rue de Sèvres
75743 PARIS Cedex 15
Tel. 01 44 49 40 00

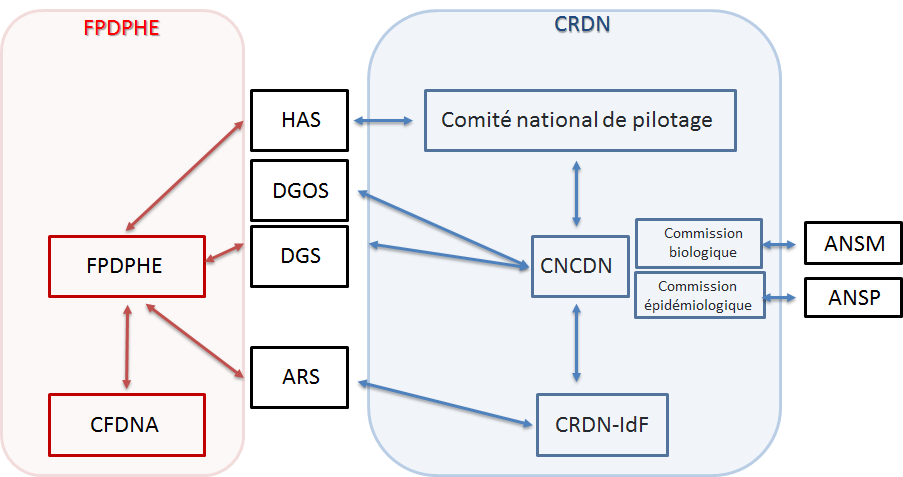
- ANSM : Agence Nationale de Sureté du Médicament
- ANSP : Agence Nationale de Santé Publique
- ARS : Agence Régionale de Santé
- CFDNA : Coordination Francilienne de Dépistage Néonatal de l’Audition
- CNCDN : Centre National de Coordination du Dépistage Néonatal
- CRDN : Centre Régional de Dépistage Néonatal
- DGS : Direction Générale de la Santé
- DGOS : Direction Générale de l’Offre de Soins
- FPDPHE : Fédération Parisienne pour le Dépistage et la Prévention des Handicaps de l’Enfant.
Arrêté du 22 février 2018, du 12 novembre 2020 et du 9 novembre 2022 relatif à l’organisation du programme national de dépistage néonatal recourant à des examens de biologie médicale
- Identifier et former des professionnels de santé responsables du prélèvement.
- S’assurer de la réalisation du prélèvement à J3 et de l’envoi du buvard par voie postale (non soumis à l’ADR), sous enveloppe T prépayée (identifiable par son bandeau rouge) dans les délais.
- Réceptionner et enregistrer les buvards ; vérifier la réception des buvards de tous les nouveau-nés par rapport aux listes d’exhaustivité des naissances transmises par les maternités.
- Vérifier la conformité des buvards, des conditions pré-analytiques requises.
- Assurer la qualité du traitement analytique, et valider les résultats selon les algorithmes nationaux.
- S’assurer de la communication des résultats anormaux du DNN aux médecins pour la convocation des nouveau-nés suspectés pour une maladie.
- S’assurer de la prise en charge et de la confirmation diagnostique du nouveau-né suspect dans les délais, en lien avec les structures de soins et les pédiatres référents pour chaque maladie dépistée.
- Archiver les résultats en assurant la confidentialité des informations.
- Conserver les buvards dans les délais réglementaires de 1 an entre 2 et 8°C.
- Surveiller régulièrement par trimestre les indicateurs qualité de la réalisation du DNN et transmettre les résultats au centre national de coordination du dépistage néonatal (CNCDN).
- Rédiger le rapport annuel d’activité du CRDN-IdF et le transmettre au CNCDN.
- Communiquer le rapport annuel d’activité du CRDN-IdF aux autorités régionales administratives de santé (ARS Île-de-France) et aux autorités nationales de santé (DGOS, DGS) au sein du Ministère de la Santé, qui assurent le financement du DNN.
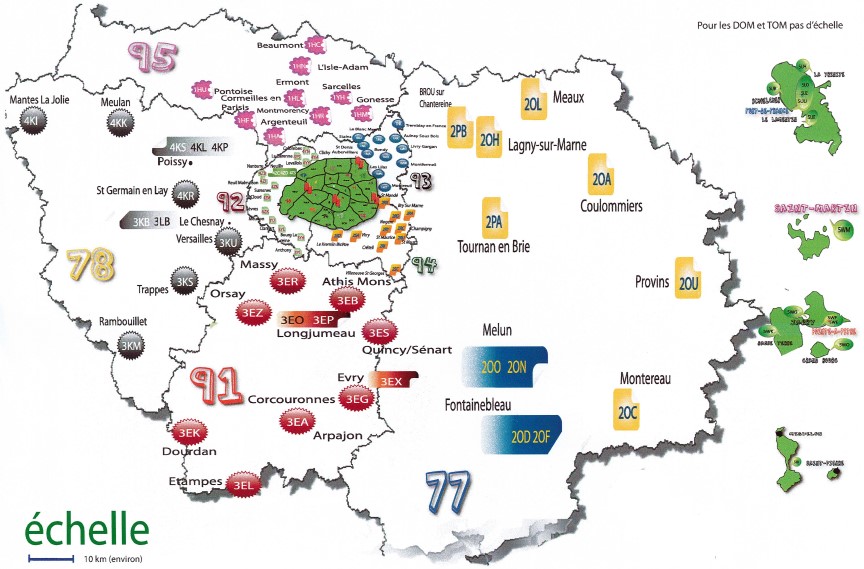
Le dépistage néonatal est mis en œuvre sur tout le territoire français au sein des centres régionaux de dépistage néonatal (CRDN). En 2018, 12 CRDN couvrent le territoire de la métropole et 5 pour le territoire des régions ultra-marines. Le centre régional d’Île-de-France réalise le dépistage biologique pour l’Île-de-France, la Martinique, la Guadeloupe, St Pierre et Miquelon et la Polynésie Française, ce qui représente près de 25% de l’activité nationale.
Les CRDN travaillent en lien étroit avec le CNDCN qui est porté par le CHU de Tours depuis le 1er juillet 2018 par l’arrêté 2017-OS-0081. Le CNDCN assure l’interface entre les différentes instances nationales (DGOS, DGS) et régionales (ARS) du dépistage néonatal. Il a pour objet de faciliter, accompagner et suivre la mise en œuvre du DNN dans les meilleures conditions possibles, contribuant ainsi à en assurer l’exhaustivité, l’homogénéité et la qualité sur le territoire national à l’aide de 2 commissions, biologique et épidémiologique.
Le centre régional d’Île-de-France présente une structure fédérative au sein de l’hôpital Necker-Enfants malades – AP-HP.Centre Université de Paris, comprenant une unité fonctionnelle médicale dépendante du département médico-universitaire (DMU) médecine de l’enfant et de l’adolescence et une UF biologique dépendante du DMU BioPhyGen.
Le coordonnateur du centre régional d’Île-de-France est le Pr Michel Polak, professeur des universités-praticien hospitalier (PU-PH) et chef de service du service d’endocrinologie, gynécologie et diabétologie pédiatriques. Il exerce également des activités de recherche au sein de l’unité Inserm U1016 et à l’Institut Imagine. Il anime et fait partie des centres de référence de maladies rares : centre de référence des pathologies gynécologiques rares, des maladies endocriniennes rares de la croissance et du développement. Il est membre de l’ENDO-ERN (European Reference Network-Endocrinology), réseau européen de soins en endocrinologie.
Le Dr Thao Nguyen Khoa est PH référente biologique pour le centre régional d’Île-de-France . Elle est rattachée au centre de référence maladies rares : mucoviscidose et affections liées à CFTR (site constitutif), et participe aux activités transversales au sein de l’unité Inserm U1151 (mucoviscidose et autres maladies épithéliales respiratoires par défaut de repliement protéique) de l’Institut Necker-Enfants malades.
Le Dr Ketty Lee est PH médecin biologiste. Elle est plus particulièrement responsable de l’activité de génétique de la mucoviscidose. Elle est également affiliée à la filière maladies constitutionnelles rares du globule rouge et de l’érythropoïèse (MCGRE).
La coordinatrice administrative est Sandrine Lachambre, cadre supérieur de santé. Elle assure la coordination en lien avec une centaine de maternités de la région Île-de-France, de la Martinique, la Guadeloupe, St Pierre et Miquelon et la Polynésie Française. Elle effectue avec l’équipe médicale du CRDN-IdF le suivi des nouveau-nés suspects afin de s’assurer de leur prise en charge. Elle coordonne les activités avec le CNCDN, les ARS (IdF-Antilles-PF) et la direction de la santé de la Polynésie Française. Elle collabore avec les directions au sein de l’hôpital Necker-Enfants malades. Mme Lachambre est en relation avec Mme Valérie Gauthereau, directrice de la CFDNA pour le dépistage de l’audition.
Centre régional de dépistage néonatal – Île-de-France
- Pr Michel Polak : PU-PH, pédiatre, coordonnateur
- + 1 pédiatre
- Sandrine Lachambre : Coordinatrice administrative/Cadre supérieure de Santé
- + 7 secrétaires médicaux
- + 7 techniciens de laboratoire médical
- Dr Thao Nguyen-Khoa : PH, biologiste référent
- + 1 biologiste PH
- Dr Ketty Lee : PH, biologiste généticienne
Autres laboratoires impliqués
- Laboratoire de Génétique moléculaire, Hôpital Necker-Enfants malades – AP-HP.Centre Université de Paris
Responsable : Dr Julie Stefann - Laboratoire de biochimie-hormonologie, UF de Dépistage Néonatal de la Drépanocytose, Hôpital Robert-Debré AP-HP Nord Université de Paris
Responsable : Dr Bichr Allaf - Laboratoire de biologie médicale, Centre hospitalier Delafontaine, Saint-Denis
Responsable : Dr Fatima Kaddari-Himeur
Coordination francilienne du dépistage néonatal de l’audition
- Valérie Gauthereau : Directrice
- +1 Secrétaire médicale
Définition
Le dépistage consiste à repérer dans une population, en l’absence de tout risque ou symptôme individuel identifié, les personnes porteuses d’une maladie.
Le DNN n’a d’intérêt que s’il permet la mise en place précoce d’un traitement efficace, modifiant ainsi le cours de l’évolution de la maladie, limitant ou empêchant l’apparition de lésions irréversibles. Il ne peut concerner que des maladies graves, repérables à la naissance par un marqueur biochimique sensible et spécifique et qui peut être dosé par une technique performante et peu coûteuse. Le DNN ne peut a contrario s’appliquer à des maladies non traitables ou pour lesquelles un marqueur biochimique détectable à la naissance et fiable n’est pas encore disponible.
Pourquoi
L’intérêt coût/bénéfice pour la population générale doit être démontré. Cette stratégie de dépistage universel et systématique de tous les nouveau-nés, financée par l’Assurance Maladie, permet un diagnostic avant la survenue des manifestations cliniques chez l’enfant et donc d’éviter ou de limiter les complications sévères dues à un diagnostic plus tardif. La réflexion pour l’introduction d’autres maladies dans le DNN est permanente et dépend de l’évolution des connaissances et des technologies. La liste des maladies faisant l’objet d’un DNN en application de l’article R1131-21 du Code de la Santé Publique est fixée par le Ministre de la Santé après avis de l’Agence de le Biomédecine (arrêté du 22 janvier 2010).
1. Les maladies endocriniennes
Hypothyroïdie congénitale
L’hypothyroïdie congénitale (HC) est une maladie liée à une sécrétion insuffisante d’hormones thyroïdiennes par la glande thyroïde. Elle touche environ un enfant sur 3 000, et elle est la principale cause de retard mental évitable. Son incidence est en augmentation en France,1/2 897 en 2015 et 1/2 631 en 2021
Dans 67% des cas l’hypothyroïdie congénitale est provoquée par une dysgénésie thyroïdienne, soit par défaut de migration pendant le développement embryonnaire, soit par hypoplasie ou agénésie thyroïdienne. Les 33% restants sont dus à un trouble de l’hormono-synthèse. Il existe aussi de très rares cas d’hypothyroïdie congénitale d’origine centrale (hypophysaire ou hypothalamique).
Le marqueur biologique de cette maladie est la TSH. Son dosage par immuno-fluorescence, permet de diagnostiquer l’hypothyroïdie congénitale précocement et d’instaurer un traitement par L-thyroxine avant que l’enfant présente des symptômes de la maladie. Le traitement instauré dans les deux premières semaines de vie permet à l’enfant de développer tout son potentiel intellectuel ; cela justifie donc un dépistage néonatal. Suite au diagnostic d’une hypothyroïdie congénitale, l’enfant sera suivi par un endocrinologue pédiatre, qui instaurera et adaptera le traitement, qui devra être administré une fois par jour, par voie orale, toujours à la même heure, afin de minimiser les éventuels oublis.
>> Stoupa A et al. Dépistage néonatal des endocrinopathies. Endocrinologie Pédiatrique 2022 ; Les dix points clés en Endocrinologie pédiatrique : chapitres 5 : p 39-44.
Hyperplasie congénitale des surrénales
L’hyperplasie congénitale des surrénales (HCS) est due à un trouble de fonctionnement des glandes surrénales.
Les glandes surrénales sont situées au-dessus des reins, et produisent plusieurs hormones importantes : le cortisol (glucocorticoïde) qui joue un rôle important dans le métabolisme du sucre et dans la lutte anti-fatigue et le stress, et l’aldostérone (minéralocorticoïde) qui intervient dans la régulation des mouvements de l’eau et du sel dans le corps. Elles sécrètent également en faible quantité des hormones mâles (testostérone).
L’hyperplasie congénitale des surrénales est le plus souvent liée au déficit de l’enzyme 21-β-hydroxylase qui entraîne une diminution de production d’aldostérone et de cortisol, et une accumulation d’hormones précurseurs, telles que la 17-hydroxy-progestérone (17OHP), ce qui conduit à une fabrication excessive de testostérone et d’autres hormones mâles. Cette affection qui touche en théorie un nouveau-né sur 15 000 peut être à l’origine d’une déshydratation par perte de sel dans les premières semaines de vie, qui justifie le dépistage néonatal, et d’anomalies des organes génitaux externes constatées à la naissance chez une petite fille.
Une augmentation des concentrations sanguines de 17OHP dosées par immuno-fluorescence, permet de suspecter une hyperplasie congénitale des surrénales. Compte tenu du déficit hormonal en aldostérone et en cortisol, il sera prescrit à l’enfant deux hormones : l’hydrocortisone (glucocorticoïde) et fludrocortisone (minéralocorticoïde). On ajoute aussi du sel (chlorure de sodium, NaCl) sous forme de gélules ou de sachets. Ce traitement doit être adapté à chaque enfant, et il doit être quotidien, en 2 ou 3 prises, à des horaires réguliers. La dose des traitements est ajustée par l’endocrinologue pédiatre en fonction de l’examen clinique (poids, taille, développement pubertaire) et des contrôles biologiques (natrémie, kaliémie, 17OHP, rénine, delta-4 androstènedione, testostérone). En cas de besoins accrus, il faut augmenter la dose de l’hydrocortisone, en doublant ou triplant la dose habituelle.
Un traitement hormonal adapté permet de faire face à ces troubles et d’en éviter les conséquences. Il doit être mis en œuvre dès la naissance.
>> Houang M et al. Analysis of a pitfall in congenital adrenal hyperplasia newborn screening: evidence of maternal use of corticoids detected on dried blood spot. Endocr Connect. 2022 Jun 15;11(6):e220101.
>> Stoupa A et al. Dépistage néonatal des endocrinopathies. Endocrinologie Pédiatrique 2022 ; Les dix points clés en Endocrinologie pédiatrique : chapitres 5 : p 39-44.
2. Drépanocytose
Drépanocytose
La drépanocytose est une maladie héréditaire, due à une anomalie de l’hémoglobine (Hb), protéine contenue dans le globule rouge. Les personnes atteintes souffrent, en général, d’anémie chronique et de crises douloureuses, et elles sont plus sensibles aux infections. La transmission se produit lorsque les deux parents sont porteurs du gène de la maladie, même s’ils ne sont pas malades (porteurs sains).
En France, l’Île-de-France est la région la plus touchée (hors DOM-TOM) avec 1 nouveau-né atteint sur 411 naissances. Cette maladie touche principalement les populations d’origine méditerranéenne, africaine et antillaise.
En cas de DNN positif par détection de l’HbS par spectrométrie de masse MALDI-TOF (Matrix Assisted Laser Desorption/Ionization–Time-of-Flight) ou par chromatographie liquide haute performance échangeuse de cations (CLHP-EC), une vérification est réalisée sur le même buvard soit par CLHP-EC soit par isoélectrofocalisation en gel d’agarose. Les parents et le nouveau-né seront convoqués pour réaliser des tests afin de confirmer le diagnostic et organiser la prise en charge du nouveau-né dans un Centre de Référence Maladies Rares ou un Centre de Compétences. Le dépistage permet donc de mettre en route tôt un suivi et une prévention adaptés. Les parents seront également informés du risque de maladie pour leurs autres enfants.
3. Mucoviscidose
Mucoviscidose
La mucoviscidose (ou fibrose kystique « Cystic Fibrosis » CF) est la maladie génétique polyviscérale la plus fréquente dans la population caucasienne, et touche un enfant sur 3 500 environ en France. Elle est de transmission autosomique récessive. Elle est liée à un dysfonctionnement de la protéine CFTR (Cystic Fibrosis Transmembrane conductance Regulator), qui assure le transport de l’ion chlorure à travers la membrane apicale des cellules épithéliales, situées notamment à la superficie des bronches et des canaux pancréatiques, mais aussi au niveau d’autres organes (glande sudoripare, hépatiques, canaux déférents, etc…). La sueur trop riche en chlorures laisse une sensation salée au baiser donné à l’enfant.
La fonction correcte de CFTR permet un bon équilibre hydro-électrolytique de chaque côté de la membrane. La bonne hydratation des sécrétions muqueuses permet le battement des cils vibratiles à la surface des cellules épithéliales bronchiques qui ramène en permanence à l’extérieur les impuretés et microorganismes inhalés vers l’extérieur. Quand la protéine CFTR est altérée, les sécrétions deviennent visqueuses, épaisses (maladie du mucus épais) et la clairance muco-ciliaire diminuée. Ceci conduit à un cercle vicieux entre obstruction – inflammation – colonisation bactérienne – fibrose pulmonaire, favorisant l’évolution vers l’insuffisance respiratoire.
Parfois, la mucoviscidose peut se révéler dès la naissance par une occlusion intestinale néonatale (iléus méconial) qui nécessite un traitement chirurgical ou médical (lavements). L’hypotrophie pondérale peut être présente à la naissance. Les enzymes digestives sécrétées par le pancréas n’arrivent plus à l’intestin et la digestion se fait mal. L’enfant peut avoir de la diarrhée, et la prise de poids est insuffisante.
Au niveau du pancréas, l’obstruction des canaux s’accompagne de l’augmentation dans le sang des enzymes pancréatiques, en particulier la trypsine, responsables de la digestion. Le dosage de la Trypsine ImmunoRéactive (TIR) par immuno-fluorescence, permet de suspecter la maladie. Mais l’élévation de la TIR n’est pas spécifique de la mucoviscidose. Pour rendre le dépistage plus spécifique, la recherche génétique de mutation du gène CFTR sera réalisée si le consentement des parents a été signé sur le buvard à la maternité. En cas de dépistage positif de la mucoviscidose, les parents seront convoqués dans un CRCM pour réaliser le test de la sueur, test biologique le plus sensible et le plus spécifique de la maladie.
La prise en charge nutritionnelle (à base d’extraits pancréatiques) et respiratoire (kinésithérapie, antibiothérapie adaptée en cas de besoin) réalisée précocement avec un suivi rigoureux améliorera la qualité de vie de l’enfant atteint, avec un allongement non négligeable de son espérance de vie.
À ce jour, des traitements spécifiques en fonction des mutations de CFTR présentes chez le patient sont proposées pour potentialiser les capacités de la protéine CFTR défaillante (Ivacaftor, Kalydeco®) et/ou pour corriger CFTR (Lumacaftor/Ivacaftor, Orkambi® ; Tezacaftor/Ivacaftor, Symdeko® ; à venir la trithérapie (USA octobre 2019) Elexacaftor/Tezacaftor/Ivacaftor, Trikafta®) avec des bénéfices pour le malade en termes de capacité ventilatoire, d’index de masse corporelle et de qualité de vie. Le dépistage précoce permet de mettre en route un suivi et un traitement adapté, améliorant l’état de santé des enfants dépistés.
4. Les maladies héréditaires du métabolisme
Phénylcétonurie
La phénylcétonurie est une maladie métabolique qui touche environ un nouveau-né sur 16 000. Elle est liée à une incapacité, totale ou partielle, de l’organisme à transformer un des constituants des aliments, la phénylalanine, en ses produits dérivés. Cette anomalie entraîne une accumulation de phénylalanine dans le sang, ce qui est nocif pendant la période de développement du cerveau. En absence de traitement précoce, et donc de dépistage néonatal, ceci conduit à un retard mental et des problèmes neurologiques, mais avec un régime spécifique dès les premières semaines de vie le développement sera normal.
La phénylalanine est un acide aminé essentiel, constituant de tous les aliments protéiques, surtout d’origine animal. Une quantité minimale quotidienne est nécessaire. Le régime de ces enfants doit donc apporter au bébé la stricte quantité dont il a besoin, sans excès mais sans carence. La quantité «tolérable» pour chaque patient est déterminée par ajustements successifs du régime. Le régime est strict notamment jusqu’à l’âge de 8 ans, avec un objectif de phénylalaninémie entre 3 à 5 mg/dL (soit 180 à 300 µmol/L). Après cet âge, le régime peut être un peu plus libre, mais en tenant compte de la nécessité de maintenir une concentration sanguine de phénylalanine inférieure à 20 mg/dL (soit 1200 µmol/L).
Leucinose
La leucinose (ou maladie des urines à odeur de sirop d’érable, Mapple Syrup Urinary Disease (MSUD)) est causée par des mutations de gènes codant 3 des sous-unités du complexe de la déshydrogénase des alpha-cétoacides à chaîne ramifiée (BCKDH). Ainsi, ces mutations entraînent une accumulation dans le sang des acides aminés à chaîne ramifiée (AACR, en particulier la leucine) et de leurs alpha-cétoacides correspondants. En Europe, cette maladie touche 1 à 4 nouveau-né(s) sur 250 000.
La forme MSUD classique (activité enzymatique résiduelle <2%) se manifeste dès 6-7 jours de vie par des difficultés d’alimentation en lien avec une somnolence. Une encéphalopathie associant une léthargie, des mouvements stéréotypés (dits de « boxing » et de « bicyclette ») s’installe progressivement, et en l’absence de prise en charge médicale spécialisé, un coma profond et un décès surviendront au cours des jours suivant. Ce risque de coma, potentiellement fatal, peut récidiver à tout âge de la vie, à l’occasion d’un stress catabolique comme une infection ou un jeûne prolongé, et s’installe sur quelques heures précédés de symptômes d’intoxication tels que des vomissements, une altération de la conscience, une ataxie et une dystonie aiguë chez le nourrisson, et des hallucinations, une hyperactivité, une ataxie et une pseudo-ébriété chez l’enfant et l’adulte.
Une MSUD classique chez un nouveau-né constitue une urgence médicale. La phase aiguë de la prise en charge nécessite un renforcement agressif de l’anabolisme protéique par des apports caloriques important (utilisation de glucose et de lipides intraveineux) et d’un mélange d’acides aminés sans AACR associé à une supplémentation en isoleucine et en valine. Une hémodialyse est souvent nécessaire. Une fois le nourrisson stabilisé, des apports en leucine seront réintroduits par des quantités limités de lait maternisé 1er âge, associé à des produits diététiques complétant les apports en acides aminés (mais sans AACR) en calories, vitamines et minéraux. La surveillance ambulatoire sera étroite avec des dosage sanguin hebdomadaires des AACR, et le suivi médical et diététique sera assuré par un centre de référence en maladies héréditaires du métabolisme. Le pronostic est favorable lorsque le patient est pris en charge de manière précoce et intensive, et lorsque le suivi permet d’éviter de nouveaux comas et maintient les concentrations plasmatiques de leucine plasmatique entre 2 et 5 mg/dL.
Tyrosinémie de type 1
La tyrosinémie de type 1 (HT-1) est une maladie héréditaire du métabolisme des acides aminés caractérisée par une atteinte hépatorénale. Sa transmission est autosomique récessive. Elle est due à un déficit en fumaryl acétoacétase (FAH) dans la voie de dégradation de la tyrosine, se traduisant par l’inhibition d’une enzyme clé de la synthèse des porphobilinogènes (la delta amino lévulinate dehydratase). La maladie touche en Europe 1 à 4 nouveau-né(s) sur 250 000.
Dans la forme infantile précoce aiguë, l’insuffisance hépato-cellulaire s’installe et devient symptomatique entre 15 jours et 3 mois de vie, avec vomissements, diarrhée, ictère, hypoglycémie, œdème, ascite et syndrome hémorragique. S’y associent une tubulopathie. La maladie peut parfois s’exprimer plus tardivement dans l’enfance par un rachitisme vitamino-résistant lié à la tubulopathie et une cirrhose. Non traitée, la maladie peut conduire au décès par des complications de l’insuffisance hépato-cellulaire aiguë, ou se compliquer de crises neurologique pseudo porphyriques. Si le traitement a été débuté tardivement, après l’âge de 6 mois, ou mal suivi, le risque d’apparition d’un carcinome hépatocellulaire au cours de l’évolution est important.
Le traitement repose sur le médicament Nitisinone (NTBC) en association avec un régime limité en protéines pour éviter l’hypertyrosinémie, et la surveillance de l’imagerie hépatique et de l’alpha fœtoprotéine marqueur sanguin sensible de l’hépatocarcinome. Ce cancer requière une transplantation du foie.
Grâce au dépistage néonatal, la mortalité et le risque de carcinome hépatique vont diminuer.
Homocystinurie classique
La maladie est une anomalie héréditaire du métabolisme de la méthionine transmise sur le mode autosomique récessif. Elle est causée par des mutations du gène CBS qui code pour l’enzyme Cystathionine bêta-synthase (CBS) qui métabolise l’homocystéine en cystathionine par la voie de trans-sulfuration du cycle de la méthionine, à l’aide du cofacteur pyridoxal 5-phosphate. Les 2 autres cofacteurs impliqués dans la voie de re-méthylation de la méthionine sont la vitamine B12 et l’acide folique.
Les manifestations cliniques sont en rapport avec l’élévation importante de l’homocystéine totale dans le plasma. Sans traitement, la maladie est progressive et se manifeste par des ectopie des cristallins et un décalage des acquisitions dans l’enfance (85% des cas) et des anomalies squelettiques (cyphose, scoliose, ostéoporose) plus tard dans la vie. Des épisodes thrombo-emboliques sévères (AVC, infarctus du myocarde, embolies pulmonaires) peuvent mettre en jeu le pronostic vital.
Si la maladie est diagnostiquée chez le nouveau-né, le but du traitement est d’assurer son bon développement et de prévenir l’apparition des complications.
Certains patients ont une forme dite sensible à la pyridoxine (Type I), puisque ce traitement permet chez eux et à lui seul de réduire très fortement le niveau d’homocystéine totale. Un supplément en acide folique et en vitamine B12 et parfois nécessaire.
Chez les sujets non répondant à la pyridoxine (Type II, pyridoxine résistant), le traitement recommandé associe alors régime pauvre en méthionine (donc hypoprotidique), acide folique, vitamine B12 et bétaïne anhydre, cette dernière permettant de réduire le taux d’homocystéine en la reméthylant en méthionine.
Grâce au dépistage néonatal, la prise en charge très précoce va conduire à la réduction des complications, notamment thrombo-emboliques, à la prévention du retard psychomoteur.
Déficit MCAD (Medium-Chain-Acyl-CoA-Déshydrogènase)
Le déficit en acyl-CoA déshydrogénase des acides gras à chaines moyennes (Medium-Chain-Acyl-CoA-Déshydrogénase, MCADD) est le plus fréquent des déficits de la bêta-oxydation des acides gras. Ce déficit n’entraine aucune manifestation pendant la vie intra-utérine. Les nouveau-nés atteints sont normaux à la naissance. La grande majorité restent asymptomatiques. Ce déficit entraîne cependant une difficulté de l’organisme à utiliser les graisses comme source d’énergie : les conséquences pour l’enfant peuvent être très graves lorsque les besoins en énergie dépassent ce que son corps réussit à produire, notamment en cas d’infection, de vomissement ou de périodes pendant lesquelles il ne s’alimente pas assez. Dans ces situations, l’enfant déficitaire en MCAD risque de décompenser en présentant des symptômes tels qu’une hypoglycémie sans cétose, des troubles du rythme cardiaque et, éventuellement, une mort subite. Le risque de décompensation devient minime à l’âge adulte. Des conseils sur l’alimentation (prévention du jeûne) et des mesures médicales au cours des situations à risque de décompensation préviennent les décompensations graves de la maladie. Ils visent à éviter les périodes de jeûne et à assurer des apports en glucides suffisants en toute circonstance. Ces mesures permettent aux enfants diagnostiqués de se développer normalement.
Le dépistage du déficit en MCAD est mis en place à partir du 1er décembre 2020 en France.
La fréquence estimée est d’environ 1/26 000 bébés.
Déficit LCHAD
Autre déficit de la bêta-oxydation des acides gras, le déficit en déshydrogénase des hydroxyacyl-CoA des acides gras à chaine longue (Long-Chain 3-hydroxyacyl-CoA-Dehydrogenase deficiency, LCHADD) touche en Europe, en moyenne un nouveau-né sur 100 000. Les premiers symptômes apparaissent entre la naissance et l’âge de 2 ans, à l’occasion d’un jeune ou d’une infection. Ces contextes conduisent à une décompensation métabolique qui peut associer : hypoglycémie sans cétose, acidose, hypotonie, troubles neurologiques pouvant aller jusqu’au coma, insuffisance hépatocellulaire, troubles du rythme cardiaque, rhabdomyolyse. A côté de ces décompensations, à partir de l’adolescence peuvent apparaître des symptômes chroniques tels qu’une neuropathie périphérique, une rétinopathie ou une myopathie. Le traitement consiste à introduire un régime très limité en acides gras à chaine longue et de les substituer par des acides gras à chaine moyenne. L’enfant doit se nourrir régulièrement et éviter le jeûne, à cet effet une nutrition entérale nocturne peut être proposée chez certains enfants. Un régime pour situation d’urgence doit être connu des parents et disponible rapidement, il permettra le plus souvent d’éviter une décompensation.
Grâce au dépistage néonatal, la survie à 10 ans des enfants malades devrait passer de 14.3% (diagnostic tardif) à 65%.
Déficit de la captation de la carnitine libre
Autre déficit de la bêta-oxydation des acides gras, le déficit systémique primaire en carnitine (Carnitine Uptake Deficiency, CUD) touche en Europe en moyenne, un nouveau-né sur 200 000. Une des étapes du métabolisme énergétique est constituée par le transfert mitochondrial des acides gras à chaînes longues et la carnitine permet ce transfert via un transporteur spécifique. La carnitine est donc essentielle pour le bon fonctionnement des muscles, du cœur, des reins et d’autres organes. Il existe plusieurs tableaux cliniques d’entrée dans la maladie :
– la forme infantile, métabolique, entre l’âge de 3 mois à 2 ans : elle est déclenchée par le jeûne ou des épisodes fébriles. La décompensation s’accompagne d’hypoglycémie sans cétose, de coma avec hyperammoniémie, cytolyse hépatique, hépatomégalie. Les crises peuvent laisser des séquelles neurologiques qui se traduiront par un retard de développement moteur et cognitif. Les enfants plus âgés peuvent présenter une cardiomyopathie progressive avec ou sans faiblesse musculaire. En l’absence de traitement, des évènements de type dyspnée, œdème cérébral, convulsions, troubles du rythme cardiaque peuvent conduire au coma et au décès.
– la forme cardiaque de l’enfant (1 à 7 ans) : insuffisance cardiaque progressive avec cardiomyopathie dilatée, hypotonie, myolyse.
– la forme adulte, rare : cardiomyopathie dilatée, troubles du rythme aiguë, fatigabilité, myopathie. L’expression clinique est très variable et peut dans certains cas être totalement asymptomatique. Cependant, les décompensations peuvent survenir à tout moment et être fatales (mort subite par troubles du rythme cardiaque).
La prise en charge consiste à administrer de la L-carnitine (Levocarnil®) par voie orale, à vie. Le pronostic est excellent tant que le patient prend son traitement. Tout arrêt de traitement entraine en quelques semaines une déplétion de l’organisme en carnitine, pouvant conduire à des manifestations aigues graves (troubles du rythme cardiaque, mort subite).
Acidurie glutarique de type 1
Le déficit en glutaryl-CoA déshydrogénase est une maladie neuro-métabolique, autosomique récessive. Elle touche en Europe 1 nouveau-né sur 100 000. Elle est due à des mutations du gène GCDH impliqué dans les voies cataboliques de la L-lysine, la L-hydroxylysine et le L-tryptophane. L’absence ou l’insuffisance de fonctionnement de l’enzyme entraine l’accumulation d’acide glutarique et 3-hydroxyglutarique au niveau cérébral.
La forme infantile « classique » (80 à 90% des cas) débute souvent entre 3 et 36 mois, brutalement, par une détresse neurologique aiguë, survenant le plus souvent pendant un épisode fébrile ou de jeûne. Elle se traduit par des signes neurologiques aigus avec un risque de handicap lourd (dystonie, retard psychomoteur…), voire un risque vital.
La prise en charge quotidienne de la maladie comprend un régime faible en lysine et tryptophane, une supplémentation en carnitine, associés à un protocole d’urgence en cas de maladie intercurrente pour éviter une décompensation neurologique aiguë. Les facteurs déclenchants de ces décompensations aiguë doivent être connus du patient et de sa famille : fièvre, jeûne prolongé, prise de poids insuffisante chez un nourrisson, pathologie intercurrente, etc. Dans ces situations, il est important de procéder à une augmentation de l’apport énergétique (viser 120% des apports journaliers recommandés), à la suppression des apports en protéines pendant 24-48h, et d’un doublement de la supplémentation en L-carnitine. L’observance des recommandations de traitement d’urgence est impérative pour prévenir les dommages neuronaux et la dystonie secondaire résultante.
Acidurie isovalérique
L’acidémie isovalérique (IVA) est une maladie autosomique récessive due à des mutations du gène IVD codant pour l’enzyme isovaléryl-CoA déshydrogénase affectant le métabolisme de la Leucine. Elle touche en Europe près de 1 nouveau-né sur 100 000.
La maladie est caractérisée par une présentation clinique variable allant d’une décompensation métabolique aiguë à début néonatal à des manifestations non spécifiques chroniques à début plus tardif, dont un retard de développement. Tous ces malades sont sujets à des épisodes intermittents de décompensation métabolique aiguë caractérisés biologiquement par des fortes concentrations urinaires de dérivés de l’acide isovalérique.
La présentation néonatale aiguë se caractérise par l’apparition, dans les deux premières semaines de vie, de vomissements, d’un état léthargique progressant vers un coma. Les examens biologiques révèlent une acidose métabolique avec augmentation du trou anionique, une hyper-ammoniémie et une cétose. En l’absence de traitement, l’évolution peut être fatale.
La maladie peut également devenir symptomatique plus tardivement dans la vie avec des signes non spécifiques, à type de retard de développement ou d’épisodes intermittents de somnolence à l’occasion d’infection intercurrentes.
Tous les patients sont susceptibles de développer des épisodes récurrents de décompensation aiguë. Les épisodes de décompensation métabolique aiguë sont déclenchés par une période prolongée de jeûne, ou suite à un apport alimentaire plus important en protéine, ou après une infection. Elles peuvent être létales en l’absence de traitement immédiat. L’odeur caractéristique de l’acide isovalérique peut être présente (similaire à l’odeur de pieds en sueur). Un retard de développement sévère et des séquelles neurologiques sont observés chez certains patients.
Une prise en charge à vie, reposant sur un régime limité en protéines, est nécessaire. Les enfants recevant un diagnostic d’IVA doivent être suivis par un médecin et un diététicien spécialisé en maladies héréditaires du métabolisme. Des compléments alimentaires à base de protéines artificielles faibles en leucine peuvent être nécessaires. La L-carnitine et la glycine peuvent être prescrites pour éliminer l’excès d’acide isovalérique. Le traitement d’urgence en cas de situation à risque de décompensation (infection, jeûne, excès d’apport alimentaire en protéine) repose sur un régime hypercalorique mais sans protéine. Cet arrêt des apports en protéine ne doit pas durer plus de 3 jours.
Le pronostic des patients diagnostiqués lors du dépistage néonatal est excellent, le développement neurologique devrait être normal si le régime et les mesures de prévention sont bien suivies.
Les patients symptomatiques peuvent conserver d’importantes séquelles neurologiques, comme un retard de développement neurologique, notamment en cas de décompensation métabolique aiguë particulièrement sévère.
Dépistage néonatal de l'audition
L’audition de l’enfant contribue à son développement global comme la vue, le toucher, le goût et l’odorat.
Les troubles auditifs peuvent être détectés dès les premiers jours de vie de l’enfant grâce à une sonde placée dans le conduit auditif (otoémissions acoustiques automatisées, OEA) ou à l’aide d’un casque posé sur les oreilles du nouveau-né (potentiels évoqués auditifs automatisés, PEAA). Le test est indolore et inoffensif pour le nouveau-né. Si une anomalie est repérée, une prise en charge rapide et adaptée est organisée dans les meilleurs délais pour éviter un retard de développement de la communication qui entrave l’épanouissement de l’enfant.

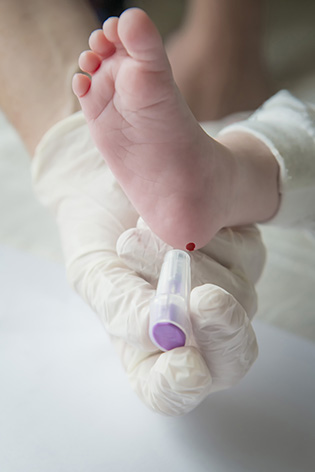
Le dépistage néonatal est réalisé en prélevant des gouttes de sang sur un buvard fourni par le CRDN, après une petite piqûre habituellement au talon du nouveau-né (ou par une prise de sang avec un petit cathlon). Il est systématiquement proposé. Les parents doivent bénéficier d’une information claire, précise, compréhensible sur le programme mis en place (le dépliant « 3 jours, l’âge du dépistage » est remis aux parents) et accepter l’acte qui leur est proposé « dépister pour prendre en charge efficacement ». Le consentement écrit (sur le buvard) est indispensable pour le dépistage génétique de la mucoviscidose. Pris en charge par l’Assurance Maladie, le prélèvement est fait le plus souvent en maternité – parfois au domicile – au plus tôt 48 heures après la naissance, au mieux après 72 heures.
Consentement génétique
Un consentement parental pour l’étude génétique est exigé. Il sera utile uniquement pour la confirmation en cas de suspicion de mucoviscidose suite à une élévation anormale du premier marqueur spécifique TIR.
Circuit des examens
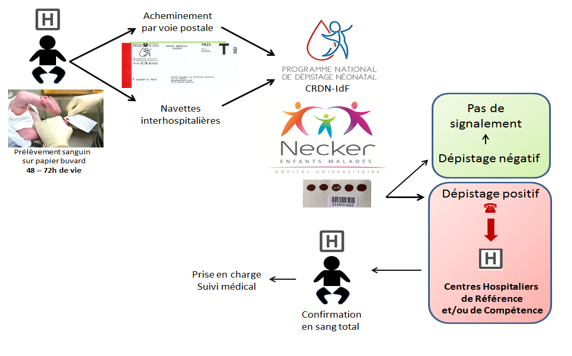
Que se passe-t-il en cas de dépistage positif ?
Un résultat de dépistage anormal doit obligatoirement être vérifié au plus vite : c’est l’étape indispensable de confirmation diagnostique. Elle est réalisée au cours de la prise en charge médicale. La consultation doit être faite par un médecin référent pour la maladie suspectée. Il reprécisera les conditions de la consultation, le dépistage et son résultat, la maladie évoquée, les tests de confirmation diagnostique qui seront pratiqués chez l’enfant.
Une fois le diagnostic de certitude acquis, le médecin référent organisera la prise en charge de la maladie, la mise en place de la thérapie spécifique, informera sur l’évolution attendue de la maladie et la surveillance proposée. Si besoin, il peut être envisagé un accompagnement psychologique et social.

Périnatalité
- AMELI > Suivi de l’enfant et de la mère après l’accouchement
- Santé publique Paris > Dépistage néonatal : la prévention commence dès la naissance
- Vidéo du prélèvement
Santé publique

Enjeux, chiffres et acteurs du dépistage néonatal en Île-de-France.

Le test de Guthrie est réalisé à la maternité entre 48h et 72h de vie du bébé, avec le consentement des parents.

Les buvards sont réceptionnés au centre régional de dépistage néonatal qui contrôle la conformité des prélèvements et de toutes les informations obligatoires.

Les prélèvements sont ensuite analysés par des appareils de spectométrie de masse au laboratoire du CRDN.

Laurence et sa fille Léa, toutes deux atteintes d’une hypothyroïdie congénitale, nous parlent du dépistage néonatal.
 Un test indolore pour repérer précocement les troubles de l’audition du nouveau-né.
Un test indolore pour repérer précocement les troubles de l’audition du nouveau-né.

Journée spéciale extension du programme national de dépistage néonatal (DNN), organisée par la filière G2M. Session de la matinée.
Journée spéciale extension du programme national de dépistage néonatal (DNN), organisée par la filière G2M. Session de l’après-midi.

Robert Guthrie est un médecin américain né à Marionville, Missouri, le 28 juin 1916, mort à Seattle le 24 juin 1995. Robert Guthrie est considéré aujourd’hui dans le monde entier comme le grand pionnier des tests de masse chez les nouveau-nés.
Son deuxième enfant était atteint de retard mental et l’une de ses nièces était atteinte de phénylcétonurie, faute d’avoir été dépistée à temps. Il a alors procédé à de nombreuses expériences qui ont enfin conduit au développement du test de Guthrie, particulièrement simple et bon marché : quelques gouttes de sang prélevées au talon ou au doigt du bébé, sur un papier absorbant.
Son test est encore aujourd’hui utilisé dans de très nombreux pays où il a été généralisé à tous les nouveau-nés. En France, le test a été présenté en 1963 et s’est généralisé vers 1980.
Textes réglementaires
- Arrêté du 22 janvier 2010 fixant la liste des maladies donnant lieu à un dépistage néonatal.
- Arrêté du 22 avril 2012 relatif à l’organisation du dépistage de la surdité permanente néonatale.
- Art R.1131-21 du décret n° 2008-321 du 4 avril 2008 relatif à l’examen des caractéristiques génétiques d’une personne ou à son identification par empreintes génétiques à des fins médicales. Arrêté du 22 janvier 2010. >> lire ici
- Arrêté du 9 novembre 2022 modifiant l’arrêté du 12 novembre 2020, modifiant l’arrêté du 22 février 2018 relatif à l’organisation du programme national de dépistage néonatal recourant à des examens de biologie médicale.
- Haute Autorité de Santé. Recommandations en Santé publique. Évaluation a priori de l’extension du dépistage néonatal à une ou plusieurs erreurs innées du métabolisme par la technique de spectrométrie de masse en tandem en population générale en France 1er volet : dépistage du déficit en MCAD. Juin 2011. >> Lire ici
De nouveaux dépistages pour traquer les maladies rares chez les nouveau-nés
France 3 | 02.03.2023
Sept nouvelles maladies ont été intégrées dans un programme national pour les nouveau-nés et cela peut sauver des vies.
> Lire la suite
Le dépistage des nouveau-nés concerne désormais 13 maladies rares, pour éviter les séquelles
Europe 1 | 16.01.2023
Le dépistage néonatal élargit sa palette de recherche à treize nouvelles pathologies, soit sept de plus depuis le 1ᵉʳ janvier. L’objectif est toujours le même : déceler le plus tôt possible des maladies rares ou sévères pour lesquelles il existe des traitements. Comment ? En multipliant le dépistage dès les premiers jours de l’enfant, comme à l’hôpital Necker à Paris.
> Lire la suite
«Une version lumineuse de la santé» : comment le dépistage néonatal change le destin des enfants
Europe 1 | 20.11.2022
Pour les 50 ans du dépistage néonatal, Europe 1 s’est entretenue avec le professeur Michel Polack, chef du service endocrinologie de l’hôpital Necker à Paris. Il met en avant cette « version lumineuse de la santé » qui permet d' »éviter les conséquences délétères de maladies » chez nombre de jeunes patients.
> Lire la suite
Le centre régional de dépistage néonatal (CRDN) – Île-de-France, c’est …
1/3 245 tests positifs concernant l’hypothyroïdie congénitale*
1/15 338 tests positifs concernant l’hyperplasie congénitale des surrénales*
1/9 925 tests positifs concernant la mucoviscidose*
1/5 624 tests positifs concernant la phénylcétonurie (et hyperphénylalaninémie modérée)*
1/411 tests positifs concernant les syndromes drépanocytaires majeurs*
* données pour l’année 2021