Regional neonatal screening center of Île-de-France (CRDN-IdF) & Coordination of neonatal hearing screening of Île-de-France (CFDNA)

![]() The national neonatal screening program (DNN) concerns all newborns born in France. It aims to detect rare, severe and most often genetic diseases on a blood sample collected on blotting paper (Guthrie) at 3 days of life and with the help of medical biology tests (defined by the order of November 9, 2022 amending the order of November 12, 2020 amending the order of February 22, 2018, relating to the organization of the national neonatal screening program using medical biology examinations).
The national neonatal screening program (DNN) concerns all newborns born in France. It aims to detect rare, severe and most often genetic diseases on a blood sample collected on blotting paper (Guthrie) at 3 days of life and with the help of medical biology tests (defined by the order of November 9, 2022 amending the order of November 12, 2020 amending the order of February 22, 2018, relating to the organization of the national neonatal screening program using medical biology examinations).
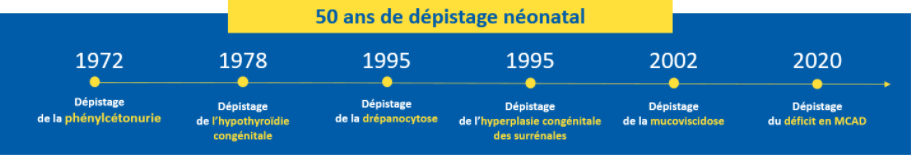
The aim of the program is to prevent the occurrence of serious manifestations and complications due to the diseases detected, at the very least to limit their severity. Because preventive therapeutic means exist, early detection brings a real benefit to newborns. Within a short period of time, it is possible to confirm the definitive diagnosis and to set up, very soon after birth, the appropriate treatment. For example, the diagnosis of phenylketonuria at birth makes it possible, with the right diet and a medical follow-up, to avoid the occurrence of severe and definitive cognitive impairment.
In 2021, the newborn screening programme in France detected :
- 284 congenital hypothyroidism of which 52 (18%) in IdF ;
- 35 congenital adrenal hyperplasia of which 9 (26%) in IdF ;
- 623 major sickle cell syndromes of which 314 (50) in IdF ;
- 118 cystic fibrosis of which 17 (14%) in IdF ;
- 46 phenylketonurias and 66 permanent moderate hyperphenylalaninemias of which 10 (22%) PKUs and 20 HPMPs (30%) in IdF ;
- Since December 1, 2020, 28 medium-chain fatty acid acyl-CoA dehydrogenase (MCAD) deficiencies of which 8 (28%) in IdF.
The introduction of tandem mass spectrometry techniques has enabled screening to be extended to other inborn errors of metabolism. As of January 1, 2023, the following diseases are included:
- 3 other aminoacidopathies : classical homocystinuria (HCY), leucinosis (MSUD) and tyrosinemia type 1 (HT-1) ;
- 2 other beta-oxidation deficiencies : long-chain 3 hydroxyacyl-CoA dehydrogenase deficiency (LCHAD) and carnitine uptake deficiency (CUD);
- 2 organic acidurias : glutaric aciduria type 1 (AG-1) and isovaleric acidemia (IVA).
The biological program is complemented by screening for permanent neonatal deafness, which is not organized by the CRDN-Île-de-France but by the perinatal networks and coordinated by the Coordination of neonatal hearing screening of Île-de-France (CFDNA), in conjunction with the ARS.
Keywords : neonatal screening, prevention, rare diseases, phenylketonuria, congenital hypothyroidism, congenital adrenal hyperplasia, sickle cell disease, cystic fibrosis, hereditary metabolic diseases.
Medical
team

Michel Polak
MD, PhD
Contact us
Necker-Enfants malades university hospital
Regional neonatal screening center
149 rue de Sèvres
75743 PARIS Cedex 15
Phone. +33 1 44 49 40 00

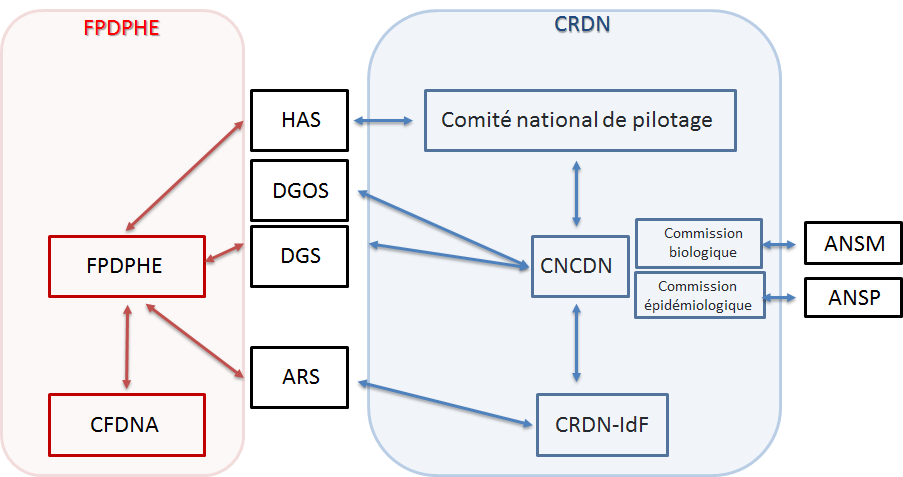
- ANSM : National Agency for the Safety of Medicines
- ANSP : National Public Health Agency
- ARS : Regional Health Agency
- CFDNA : Coordination of Neonatal Hearing Screening of Île-de-France
- CNCDN : National Coordination Center for Neonatal Screening
- CRDN : Regional Neonatal Screening Center
- DGS : General Directorate of Health
- DGOS : General Directorate of Healthcare Services
- FPDPHE : Parisian Federation for the Screening and Prevention of Childhood Disabilities.
Orders of February 22, 2018, November 12, 2020 and November 9, 2022 on the organization of the national program of neonatal screening using medical biology examinations
- Identify and train health professionals responsible for collection.
- Ensure that the sample is taken at D3 and that the blotting paper is sent by post (not subject to acknowledgement of receipt), in a prepaid T envelope (identifiable by its red banner) within the deadline.
- Receive and register blotters; check the receipt of blotters from all newborns against the birth completeness lists provided by the maternity wards.
- Check the compliance of the blotters with the required pre-analytical conditions.
- Ensure the quality of the analytical treatment, and validate the results according to national algorithms.
- Ensure the communication of abnormal neonatal screening results to doctors for the convocation of newborns suspected of a disease.
- Ensure the timely care and diagnostic confirmation of the suspect newborn, in collaboration with health care facilities and referring pediatricians for each disease detected.
- Archive the results while ensuring the confidentiality of the information
- Keep the blotters within the statutory period of 1 year between 2 and 8°C..
- Regularly monitor quarterly quality indicators of the performance of neonatal screening and transmit the results to the national neonatal screening coordination center (CNCDN).
- Write the annual activity report of the CRDN-IdF and transmit it to the CNCDN.
- Communicate the annual activity report of the CRDN-IdF to the regional administrative health authorities (ARS Île-de-France) and the national health authorities (DGOS, DGS) within the Ministry of Health, which provide funding for neonatal screening.
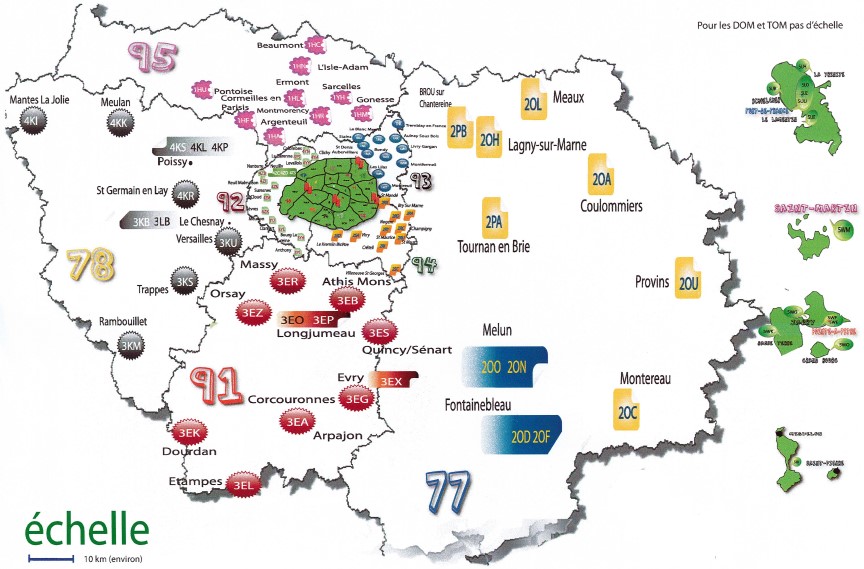
Neonatal screening is implemented throughout France in regional neonatal screening centers. In 2018, 12 regional centers cover metropolitan France and 5 for the overseas regions. The Ile-de-France regional center carries out biological screening for Ile-de-France, Martinique, Guadeloupe, St Pierre and Miquelon and French Polynesia, representing nearly 25% of national activity (2018 report).
The regional centers work in close collaboration with the national center, which is supported by the CHU of Tours since July 1, 2018 by order 2017-OS-0081. The national center acts as an interface between the different national (DGOS, DGS) and regional (ARS) authorities for neonatal screening. Its purpose is to facilitate, support and monitor the implementation of neonatal screening in the best possible conditions, thus helping to ensure its completeness, consistency and quality on the national territory with the help of 2 commissions, biological and epidemiological.
The Île-de-France regional center presents a federative structure within the Necker-Enfants malades – AP-HP.Centre University of Paris hospital, including a medical functional unit dependent on the medical-university department (DMU) child and adolescent medicine and a biological functional unit dependent on the BioPhyGen DMU.
The coordinator of the Île-de-France regional center is Pr Michel Polak, university professor – hospital practitioner (PU-PH) and head of the department of pediatric endocrinology, gynecology and diabetology. He also carries out research activities within the Inserm U1016 unit and at the Imagine Institute. He leads and is part of the reference centers for rare diseases: reference center for rare gynecological pathologies, rare endocrine diseases of growth and development. He is a member of ENDO-ERN (Endocrinology- European reference network), a European network of endocrinology care.
Dr Thao Nguyen Khoa is PH biological referent for the regional center of Île-de-France. She is attached to the reference center for cystic fibrosis and conditions linked to a CFTR abnormality and participates in transversal activities within the INSERM U1151 unit (cystic fibrosis and other respiratory epithelial diseases due to protein misfolding) at the Necker Enfants malades Institute.
Dr Ketty Lee is a PH biologist. She is in charge of the cystic fibrosis genetics activity. She is also affiliated with the rare constitutional diseases of the red globule and erythropoiesis (MCGRE) healthcare network.
The administrative coordinator is Sandrine Lachambre, senior health executive. She is in charge of coordination in connection with about a hundred maternity hospitals in the Île-de-France region, Martinique, Guadeloupe, St Pierre and Miquelon and French Polynesia. Together with the CRDN-IdF medical team, she monitors suspected newborns to ensure they are properly cared for. She coordinates activities with the CNCDN, the ARS (IdF-Antilles-PF) and the French Polynesia health department. She collaborates with the departments at Necker-Enfants malades hospital. Sandrine Lachambre is in contact with Valérie Gauthereau, director of the CFDNA for hearing screening.
Regional Neonatal Screening Center – Île-de-France
- Pr Michel Polak : PU-PH, pediatrician, coordinator
- + 1 pediatrician
- Sandrine Lachambre : Administrative coordinator/Senior health manager
- + 6 medical secretaries
- + 7 medical laboratory technicians
- Dr Thao Nguyen-Khoa : PH, referent biologist
- + 1 PH biologist
- Dr Ketty Lee : PH, genetic biologist
Other laboratories involved
- Molecular genetics laboratory, Necker hospital – AP-HP.Centre University of Paris
Person in charge: Dr Julie Stefann - Biochemistry-hormonology laboratory, sickle cell disease neonatal screening functional unit, Robert-Debré hospital AP-HP.Nord University of Paris
Person in charge: Dr. Bichr Allaf - Medical biology laboratory
Delafontaine hospital center, Saint-Denis
Person in charge : Dr Fatima Kaddari-Himeur
Île-de-France coordination of neonatal hearing screening
- Valérie Gauthereau : Director
- +1 Medical secretary
Definition
Screening consists of identifying in a population, in the absence of any identified individual risk, people who are carriers of a disease.
Neonatal screening is of interest only if early diagnosis can lead to effective treatment that can modify the course of the disease before irreversible damage occurs. It can only concern serious illnesses, which can be detected at birth by a sensitive and specific biochemical marker and which can be measured using a high-performance and inexpensive technique. On the other hand, neonatal screening cannot be applied to diseases that do not have a biochemical marker detectable at birth or that cannot be treated.
Why ?
The cost/benefit to the general population must be demonstrated. This strategy of universal and systematic screening of all newborns, financed by the Assurance Maladie, allows diagnosis before the onset of clinical manifestations in children and thus avoids or limits severe complications due to later diagnosis. Consideration for the introduction of other diseases into neonatal screening is ongoing and depends on the evolution of knowledge and technology. The list of diseases subject to neonatal screening in application of article R1131-21 of the public health code is set by the Minister of Health after advice from the biomedicine agency (order of January 22, 2010).
1. Endocrine diseases
Congenital hypothyroidism
Congenital hypothyroidism (CH) is a disease related to insufficient thyroid hormone secretion by the thyroid gland. It affects about 1 in 3,000 children and is the leading cause of preventable mental retardation. Its incidence is increasing in France, 1/2 897 in 2015 and 1/2 631 in 2021.
In 67% of cases congenital hypothyroidism is caused by thyroid dysgenesis, either due to a lack of migration during embryonic development or by thyroid hypoplasia or agenesis. The remaining 33% is due to a hormone synthesis disorder. There are also very rare cases of congenital hypothyroidism of central origin (pituitary or hypothalamic).
The biological marker for this disease is TSH. Its determination by immunofluorescence assay, allows the early diagnosis of congenital hypothyroidism and the initiation of L-thyroxine treatment before the child presents symptoms of the disease. The treatment initiated in the first two weeks of life allows the child to develop his or her full intellectual potential, thus justifying neonatal screening. Following the diagnosis of congenital hypothyroidism, the child will be monitored by a pediatric endocrinologist, who will initiate and adapt the treatment, which should be administered once a day, orally, always at the same time, in order to minimize possible forgetfulness.
>> Stoupa A et al. Dépistage néonatal des endocrinopathies. Endocrinologie Pédiatrique 2022 ; Les dix points clés en Endocrinologie pédiatrique : chapitres 5 : p 39-44.
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia (CAH) is due to a disturbance in the functioning of the adrenal glands.
The adrenal glands are located above the kidneys and produce several important hormones: cortisol (glucocorticoid) which plays an important role in sugar metabolism and in the fight against fatigue and stress, and aldosterone (mineralocorticoid) which is involved in regulating the movement of water and salt in the body. They also secrete small amounts of male hormones (testosterone).
Congenital adrenal hyperplasia is most commonly associated with a deficiency of the enzyme 21-β-hydroxylase, which leads to decreased production of aldosterone and cortisol, and an accumulation of precursor hormones, such as 17-hydroxy-progesterone (17OHP), which leads to overproduction of testosterone and other male hormones. This condition, which theoretically affects one in 15,000 newborns, can lead to dehydration due to salt loss in the first weeks of life, warranting neonatal screening, and to abnormalities of the external genitalia found at birth in a young girl.
An increase in the blood concentrations of 17OHP, assayed by immunofluorescence, makes it possible to suspect congenital adrenal hyperplasia. Due to the hormonal deficit in aldosterone and cortisol, the child will be prescribed two hormones: hydrocortisone (glucocorticoid) and fludrocortisone (mineralocorticoid). Salt (sodium chloride, NaCl) is also added in the form of capsules or sachets. This treatment must be adapted to each child, and it must be daily, in 2 or 3 doses, at regular times. The dose of the treatments is adjusted by the pediatric endocrinologist according to the clinical examination (weight, height, pubertal development) and biological controls (natremia, kalemia, 17OHP, renin, delta-4 androstenedione, testosterone). In case of increased needs, the dose of hydrocortisone should be increased, doubling or tripling the usual dose.
Appropriate hormonal treatment can cope with these disorders and avoid their consequences. It must be implemented from birth.
>> Houang M et al. Analysis of a pitfall in congenital adrenal hyperplasia newborn screening: evidence of maternal use of corticoids detected on dried blood spot. Endocr Connect. 2022 Jun 15;11(6):e220101.
>> Stoupa A et al. Dépistage néonatal des endocrinopathies. Endocrinologie Pédiatrique 2022 ; Les dix points clés en Endocrinologie pédiatrique : chapitres 5 : p 39-44.
2. Sickle cell disease
Sickle cell disease
Sickle cell disease is an inherited disease caused by an abnormality of hemoglobin (Hb), a protein contained in the red blood cell. Affected people generally suffer from chronic anemia and painful attacks, and are more susceptible to infections. Transmission occurs when both parents are carriers of the disease gene, even if they are not sick (healthy carriers).
In France, Île-de-France is the most affected region (excluding DOM-TOM) with 1 newborn reached out of 700 births. This disease mainly affects populations of Mediterranean, African and Caribbean origin.
In the event of a positive neonatal screening by HbS detection using MALDI-TOF (Matrix Assisted Laser Desorption/Ionization-Time-of-Flight) mass spectrometry or cation-exchange high-performance liquid chromatography (CE-HPLC), a check is carried out on the same blotter using either CE-HPLC or agarose gel isoelectrofocusing. The parents and the newborn will be called in for tests to confirm the diagnosis and organize care for the newborn in a rare diseases reference center or a competence center. Screening thus enables the early initiation of appropriate follow-up and prevention. Parents will also be informed of the risk of the disease for their other children.
3. Cystic fibrosis
Cystic fibrosis
Cystic fibrosis (CF) is the most common multi-organ genetic disease in the Caucasian population, and affects approximately one in 3,500 children in France. It is autosomal recessive. It is linked to a dysfunction of the CFTR protein (Cystic Fibrosis Transmembrane conductance Regulator), which ensures the transport of the chloride ion through the apical membrane of epithelial cells, located in particular on the surface of the bronchi and pancreatic ducts, but also at the level of other organs (sweat gland, hepatic, vas deferens, etc…). Sweat too rich in chlorides leaves a salty sensation when kissing the child.
The correct function of CFTR allows a good water-electrolyte balance on each side of the membrane. The good hydration of the mucous secretions allows the vibratile cilia to beat on the surface of the bronchial epithelial cells, which constantly brings the inhaled impurities and microorganisms outwards. When the CFTR protein is altered, the secretions become viscous, thick (thick mucus disease) and mucociliary clearance decreased. This leads to a vicious circle between obstruction – inflammation – bacterial colonization – pulmonary fibrosis, leading to progression to respiratory failure.
Sometimes cystic fibrosis can show itself at birth as a neonatal bowel obstruction (NBO) (meconium ileus) which requires surgical or medical treatment (enemas). Low birth weight may be present. The digestive enzymes secreted by the pancreas no longer reach the intestine and digestion is damaged. The child may have diarrhea and he/she is not getting enough weight.
In the pancreas, the obstruction of the ducts is accompanied by an increase in the blood of pancreatic enzymes, in particular trypsin, which are responsible for digestion. The assay of ImmunoReactive Trypsinogen (IRT) by immunofluorescence makes it possible to suspect the disease. But the increase in IRT is not specific to cystic fibrosis. To make the screening more specific, genetic research for a mutation in the CFTR gene will be carried out if the parents’ consent has been signed on the blotter at the maternity hospital. In the event of a positive cystic fibrosis screening, the parents will be called to a cystic fibrosis resource and competence center to perform the sweat test, the most sensitive and specific biological test for the disease.
Nutritional care (based on pancreatic extracts) and respiratory (physiotherapy, antibiotic therapy adapted if necessary) carried out early with rigorous monitoring will improve the quality of life of the affected child, with a significant extension of his expectation of life.
To date, specific treatments depending on the CFTR mutations present in the patient are proposed to potentiate the capacities of the defective CFTR protein (Ivacaftor, Kalydeco®) and / or to correct CFTR (Lumacaftor / Ivacaftor, Orkambi®; Tezacaftor / Ivacaftor, Symdeko®; forthcoming triple therapy (USA October 2019) Elexacaftor / Tezacaftor / Ivacaftor, Trikafta®) with benefits for the patient in terms of ventilatory capacity, body mass index and quality of life. Early detection makes it possible to initiate appropriate follow-up and treatment, improving the state of health of the children screened.
>> Sermet-Gaudelus I et al. Sweat chloride testing and nasal potential difference (NPD) are primary outcome parameters in treatment with Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) modulators. J Pers Med. 2021 Jul 27;11(8):729.
4. Hereditary metabolic diseases
Phenylketonuria
Phenylketonuria is a metabolic disease that affects approximately one in 16,000 newborns. It is related to a total or partial inability of the body to transform one of the constituents of food, phenylalanine, into its by-products. This abnormality leads to an accumulation of phenylalanine in the blood, which is harmful during the brain’s development. In the absence of early treatment, and therefore neonatal screening, this leads to mental retardation and neurological problems, but with a specific diet from the first weeks of life development will be normal.
Phenylalanine is an essential amino acid, constituent of all protein foods, especially of animal origin. A minimum daily amount is required. The diet of these children must therefore provide the baby with the strict quantity he needs, without excess but without deficiency. The « tolerable » quantity for each patient is determined by successive adjustments of the diet. The diet is strict in particular until the age of 8 years, with a target of phenylalaninemia between 3 to 5 mg/dL (i.e. 180 to 300 µmol/L). After this age, the diet may be slightly freer, but taking into account the need to maintain a phenylalanine blood concentration below 20 mg/dL (or 1200 µmol/L).
Maple syrup urine disease
Mapple Syrup Urinary Disease (MSUD) is caused by mutations in genes encoding 3 of the subunits of the branched-chain alpha-keto acid dehydrogenase (BCKDH) complex. These mutations lead to an accumulation of branched-chain amino acids (BCAAs, in particular leucine) and their corresponding alpha-keto acids in the blood. In Europe, this disease affects 1 to 4 out of every 250,000 newborns.
The classic MSUD form (residual enzyme activity <2%) manifests itself as early as 6-7 days of age, with feeding difficulties associated with drowsiness. An encephalopathy associating lethargy, stereotyped movements (« boxing » and « bicycling ») progressively sets in, and in the absence of specialized medical care, a deep coma and death will occur over the following days. This potentially fatal risk of coma can recur at any age, during catabolic stress such as infection or prolonged fasting, and sets in over a few hours, preceded by symptoms of intoxication such as vomiting, altered consciousness, ataxia and acute dystonia in infants, and hallucinations, hyperactivity, ataxia and pseudo-ebriety in children and adults.
Classic MSUD in newborns is a medical emergency. The acute phase of management requires aggressive reinforcement of protein anabolism with high caloric intake (use of intravenous glucose and lipids) and a mixture of amino acids without BCAA, combined with isoleucine and valine supplementation. Hemodialysis is often required. Once the infant has stabilized, leucine intake is reintroduced in limited quantities with infant formula, combined with dietary products that supplement the amino acid intake (but without BCAA) with calories, vitamins and minerals. Outpatient monitoring will be close, with weekly BCAA blood tests, and medical and dietary follow-up will be provided by a reference center for hereditary metabolic diseases. The prognosis is favourable when the patient is managed early and intensively, and when follow-up prevents further comas and maintains plasma leucine concentrations between 2 and 5 mg/dL.
Tyrosinemia type 1
Tyrosinemia type 1 (HT-1) is an inherited disease of amino acid metabolism characterized by hepatorenal involvement. It is inherited in an autosomal recessive fashion. It is due to a deficiency of fumaryl acetoacetate hydrolase (FAH) in the tyrosine degradation pathway, resulting in inhibition of a key enzyme in porphobilinogen synthesis (delta amino levulinate dehydratase). In Europe, the disease affects 1 to 4 newborns out of every 250,000.
In the acute early infantile form, hepatocellular insufficiency sets in and becomes symptomatic between 15 days and 3 months of age, with vomiting, diarrhea, jaundice, hypoglycemia, edema, ascites and hemorrhagic syndrome. Tubulopathy is associated. The disease can sometimes develop later in childhood into vitamin-resistant rickets linked to tubulopathy and cirrhosis. Untreated, the disease can lead to death from complications of acute hepatocellular failure, or be complicated by pseudoporphyric neurological crises. If treatment is started late, after the age of 6 months, or is poorly monitored, the risk of developing hepatocellular carcinoma during the course of the disease is high.
Treatment is based on the Nitisinone (NTBC) drug in combination with a protein-restricted diet to avoid hypertyrosinemia, and monitoring of liver imaging and alpha-fetoprotein, a sensitive blood marker of hepatocarcinoma. This cancer requires liver transplantation.
Thanks to neonatal screening, mortality and the risk of hepatic carcinoma will be reduced.
Classic homocystinuria
The disease is an autosomal recessive inherited disorder of methionine metabolism. It is caused by mutations in the CBS gene, which codes for the Cystathionine beta-synthase (CBS) enzyme, which metabolizes homocysteine to cystathionine via the trans-sulfuration pathway of the methionine cycle, using the pyridoxal 5-phosphate cofactor. The 2 other cofactors involved in the methionine re-methylation pathway are vitamin B12 and folic acid.
Clinical manifestations are related to the significant elevation of total homocysteine in plasma. Left untreated, the disease is progressive, manifesting itself as lens ectopia and delayed acquisition in childhood (85% of cases) and skeletal abnormalities (kyphosis, scoliosis, osteoporosis) later in life. Severe thrombo-embolic events (stroke, myocardial infarction, pulmonary embolism) can be life-threatening.
If the disease is diagnosed in a newborn, the aim of treatment is to ensure proper development and prevent the onset of complications.
Some patients have a pyridoxine-sensitive form of the disease (Type I), since this treatment alone can significantly reduce total homocysteine levels. Folic acid and vitamin B12 supplements are sometimes required.
In subjects who do not respond to pyridoxine (Type II, pyridoxine-resistant), the recommended treatment combines a low-methionine diet (i.e. hypoprotein), folic acid, vitamin B12 and anhydrous betaine, the latter of which reduces homocysteine levels by remethylating it into methionine.
Thanks to neonatal screening, very early treatment will help reduce complications, particularly thrombo-embolic ones, and prevent psychomotor retardation.
MCAD (Medium chain acyl-CoA dehydrogenase) deficiency
Medium-Chain Acyl-CoA Dehydrogenase deficiency (MCADD) is the most common deficiency of fatty acid beta-oxidation. This deficiency does not manifest itself during intrauterine life. Affected newborns are normal at birth. The vast majority remain asymptomatic. This deficiency causes the body to have difficulty using fat as an energy source.
The consequences for the child can be very serious when the child’s energy needs exceed what the body is able to produce, especially in the case of infections, vomiting or periods of underfeeding. Symptomatic newborns have hypoglycemia, heart rhythm problems and even sudden death.
Feeding advice and medical measures prevent severe manifestations of the disease. They aim to avoid periods of fasting and to ensure adequate sugar intake at all times. These measures allow diagnosed children to develop normally.
Screening for MCAD deficiency will be implemented in France as of December 1, 2020. The estimated frequency is approximately 1/26,000 babies.
LCHAD (Long-chain 3-hydroxyacyl-CoA dehydrogenase) deficiency
Another fatty acid beta-oxidation deficiency, Long-Chain 3-hydroxyacyl-CoA-Dehydrogenase deficiency (LCHADD), affects an average of one in every 100,000 newborns in Europe. The first symptoms appear between birth and the age of 2, when a young person or an infection is present. These conditions lead to metabolic decompensation, which can include hypoglycemia without ketosis, acidosis, hypotonia, neurological disorders that can even lead to coma, hepatocellular failure, cardiac rhythm disorders and rhabdomyolysis. Alongside these decompensations, chronic symptoms such as peripheral neuropathy, retinopathy and myopathy can appear from adolescence onwards. Treatment consists of introducing a diet with a very limited intake of long-chain fatty acids, and substituting them with medium-chain fatty acids. The child must eat regularly, and avoid fasting. To this end, nocturnal enteral nutrition may be proposed for certain children. Parents should be aware of the need for an emergency diet, which should be readily available to avoid decompensation.
Thanks to neonatal screening, the 10-year survival rate of sick children should rise from 14.3% (late diagnosis) to 65%.
Carnitine uptake deficiency (CUD)
Another deficiency in the beta-oxidation of fatty acids, primary systemic carnitine deficiency (Carnitine Uptake Deficiency, CUD) affects an average of one in 200,000 newborns in Europe. One of the steps in energy metabolism is the mitochondrial transfer of long-chain fatty acids, and carnitine enables this transfer via a specific transporter. Carnitine is therefore essential for the proper functioning of muscles, heart, kidneys and other organs. There are several clinical presentations of the disease:
– the infantile, metabolic form, between 3 months and 2 years of age: triggered by fasting or febrile episodes. Decompensation is accompanied by hypoglycemia without ketosis, coma with hyperammonemia, hepatic cytolysis and hepatomegaly. Seizures can lead to neurological sequelae in the form of delayed motor and cognitive development. Older children may present with progressive cardiomyopathy, with or without muscular weakness. If left untreated, events such as dyspnea, cerebral edema, convulsions and cardiac rhythm disorders can lead to coma and death.
– the cardiac form in children (aged 1 to 7): progressive heart failure with dilated cardiomyopathy, hypotonia, myolysis.
– the rare adult form: dilated cardiomyopathy, acute rhythm disorders, fatigability, myopathy. Clinical expression is highly variable, and may in some cases be totally asymptomatic. However, decompensation can occur at any time, and can be fatal (sudden death due to cardiac rhythm disorders).
Treatment consists of lifelong oral administration of L-carnitine (Levocarnil®). The prognosis is excellent as long as the patient remains on treatment. Stopping treatment within a few weeks leads to carnitine depletion in the body, which can lead to serious acute symptoms (heart rhythm disorders, sudden death).
Glutaric aciduria type 1
Glutaryl-CoA dehydrogenase deficiency is an autosomal recessive neurometabolic disease. In Europe, it affects 1 in 100,000 newborns. It is due to mutations in the GCDH gene involved in the catabolic pathways of L-lysine, L-hydroxylysine and L-tryptophan. The absence or inadequate functioning of the enzyme leads to the accumulation of glutaric and 3-hydroxyglutaric acids in the brain.
The « classic » infantile form (80-90% of cases) often begins between 3 and 36 months of age, abruptly, with acute neurological distress, most often occurring during a febrile or fasting episode. It leads to acute neurological signs, with the risk of severe disability (dystonia, psychomotor retardation, etc.), or even death.
Daily management of the disease includes a diet low in lysine and tryptophan, and carnitine supplementation, combined with an emergency protocol in the event of intercurrent illness to avoid acute neurological decompensation. The triggers for acute neurological decompensation should be known to the patient and family: fever, prolonged fasting, insufficient weight gain in infants, intercurrent pathology, etc. In these situations, it is important to increase energy intake (aiming for 120% of recommended daily intake), suppress protein intake for 24-48 hours, and double L-carnitine supplementation. Compliance with emergency treatment recommendations is imperative to prevent neuronal damage and the resulting secondary dystonia.
Isovaleric acidaemia (IVA)
Isovaleric acidemia (IVA) is an autosomal recessive disease caused by mutations in the IVD gene coding for the enzyme isovaleryl-CoA dehydrogenase, which affects leucine metabolism. In Europe, it affects around 1 in 100,000 newborns.
The disease is characterized by a variable clinical presentation, ranging from acute metabolic decompensation at neonatal onset to chronic non-specific manifestations at later onset, including developmental delay. All patients are subject to intermittent episodes of acute metabolic decompensation, characterized biologically by high urinary concentrations of isovaleric acid derivatives.
Acute neonatal presentation is characterized by the onset, within the first two weeks of life, of vomiting and a lethargic state progressing to coma. Biological tests reveal metabolic acidosis with increased anion gap, hyperammonemia and ketosis. If left untreated, the course can be fatal.
The disease can also become symptomatic later in life, with nonspecific signs such as developmental delay or intermittent episodes of drowsiness during intercurrent infections.
All patients are likely to develop recurrent episodes of acute decompensation. Episodes of acute metabolic decompensation are triggered by a prolonged period of fasting, or following increased dietary protein intake, or after an infection. They can be lethal in the absence of immediate treatment. The characteristic odor of isovaleric acid may be present (similar to the smell of sweaty feet). Severe developmental delay and neurological sequelae are observed in some patients.
Lifelong management, based on a protein-restricted diet, is required. Children diagnosed with VIA should be monitored by a physician and dietician specialized in hereditary metabolic diseases. Dietary supplements based on artificial proteins low in leucine may be necessary. L-carnitine and glycine may be prescribed to eliminate excess isovaleric acid. Emergency treatment in situations at risk of decompensation (infection, fasting, excess protein intake) is based on a high-calorie, protein-free diet. Protein intake should be stopped for no more than 3 days.
The prognosis for patients diagnosed during neonatal screening is excellent, and neurological development should be normal if diet and preventive measures are followed.
Symptomatic patients may retain significant neurological sequelae, such as delayed neurodevelopment, especially in cases of particularly severe acute metabolic decompensation.
Neonatal hearing screening
Hearing contributes to a child’s overall development, as do sight, touch, taste and smell. Hearing problems can be detected in the first days of a child’s life by means of a probe placed in the ear canal (Otoacoustic Emissions OAE) or with the help of a headset placed on the newborn’s ears (Auditory Evoked Potentials, AEP). The test is painless and harmless to the newborn. If an abnormality is detected, rapid and appropriate treatment is organized as soon as possible to avoid a delay in the development of communication that would hinder the child’s development.


![]()
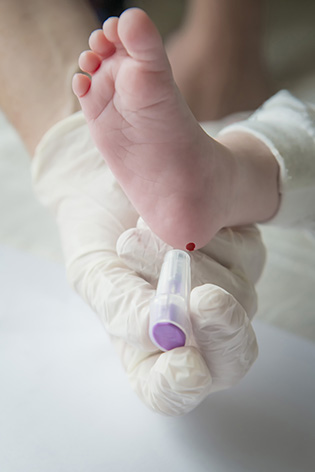
Neonatal screening is performed by taking drops of blood on a blotter provided by the CRDN, after a small puncture usually at the heel of the newborn (or by drawing blood with a small catheter). It is systematically proposed. The parents must be given clear, precise and understandable information about the program (the leaflet « 3 days, the age of screening » is given to the parents) and accept the act which is proposed to them « to screen in order to take care effectively ». Written consent (on the blotter) is essential for genetic screening for cystic fibrosis. Covered by the Assurance Maladie, the sample is usually taken in the maternity hospital – sometimes at home – at the earliest 48 hours after the birth, at best after 72 hours.
Genetic consent
Parental consent for the genetic study is required. It will only be useful for confirmation in case of suspicion of cystic fibrosis following abnormal elevation of the first specific IRT marker.
Examination path
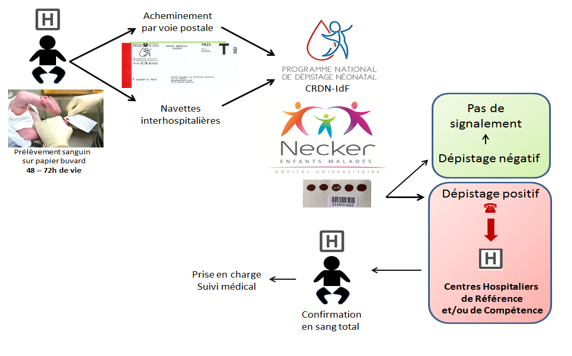
What happens if I test positive?
An abnormal screening result must be checked as soon as possible: this is the essential step of diagnostic confirmation. It is carried out during medical care. The consultation must be made by a referring physician for the suspected disease. He will repeat the conditions of the consultation, the screening and its result, the evoked disease, and the diagnostic confirmation tests that will be performed on the child.
Once the diagnosis of certainty is acquired, the referring physician will organize the care of the disease, the implementation of the specific therapy, inform on the expected evolution of the disease and the proposed surveillance. If necessary, psychological and social support may be considered.

Perinatality
- AMELI > Child and mother follow-up after childbirth
- Paris public health > Neonatal screening: prevention begins at birth
- Video of the sample
Public health
- Ministère des solidarités et de la santé > National neonatal screening program
- ARS Île-de-France > Regional specifications for the screening program for permanent bilateral neonatal deafness (SPBN)
- National neonatal screening program progress report, Year 2018
- French society of pediatrics
- Rare disease alliance
- Rare diseases and orphan drugs portal

Issues, figures and actors of neonatal screening in Île-de-France.

The Guthrie test is performed in the maternity ward between 48 and 72 hours of the baby’s life, with the parents’ consent.

The blotters are received at the regional neonatal screening center, which checks the compliance of the samples and all mandatory information.

The samples are then analyzed by mass spectrometry equipment at the CRDN laboratory.

Laurence and her daughter Lea, both with congenital hypothyroidism, talk about neonatal screening.
 A painless test for early detection of hearing problems in newborns.
A painless test for early detection of hearing problems in newborns.

Special day on the extension of the national neonatal screening program (DNN), organized by the G2M network. Morning session.
Special day on the extension of the national neonatal screening program (DNN), organized by the G2M network. Afternoon session.

Robert Guthrie was an American physician born in Marionville, Missouri, on June 28, 1916, who died in Seattle on June 24, 1995. Today, Robert Guthrie is considered worldwide as the great pioneer of mass testing of newborns.
His second child was mentally retarded and one of his nieces had phenylketonuria because she was not detected in time. He then carried out numerous experiments which finally led to the development of the Guthrie test, which is particularly simple and inexpensive: a few drops of blood are taken from the baby’s heel or finger, on absorbent paper.
Hiss test is still used today in many countries where it has been generalized to all newborns. In France, the test was introduced in 1963 and became widespread around 1980.
Regulatory texts
- Order of January 22, 2010 establishing the list of diseases giving rise to neonatal screening.
- Order of April 22, 2012 relating to the organization of screening for permanent neonatal deafness.
- Art R.1131-21 of the decree n° 2008-321 of April 4, 2008 relating to the examination of the genetic characteristics of a person or his identification by genetic fingerprints for medical purposes. Order of January 22, 2010. >> Read here
- Order of November 9, 2022 amending the order of November 12, 2020, amending the order of February 22, 2018 on the organization of the national neonatal screening program using medical biology tests.
- High Authority of Health. Recommendations in Public Health. A priori evaluation of the extension of neonatal screening to one or more innate errors of metabolism using the tandem mass spectrometry technique in the general population in France 1st part: screening for MCAD deficiency. June 2011. >> Read here
New screenings to track rare diseases in newborns
France 3 | 03.02.2023
Seven new diseases have been incorporated into a national program for newborns and this can save lives.
> Read more
Newborn screening now covers 13 rare diseases, to avoid sequelae
Europe 1 | 01.16.2023
Newborn screening is expanding its research palette to thirteen new conditions, seven more since January 1ᵉʳ. The goal remains the same: to detect rare or severe diseases for which treatments exist as early as possible. How? By multiplying screening from the child’s first days, as at the Necker hospital in Paris.
> Read more
« A bright version of health »: how newborn screening is changing the fate of children
Europe 1 | 11.20.2022
For the 50th anniversary of neonatal screening, Europe 1 spoke with Professor Michel Polack, head of the endocrinology department at Necker Hospital in Paris. He highlights this « luminous version of health » which allows to « avoid the deleterious consequences of diseases » in many young patients.
> Read more
In Necker, the regional neonatal screening center (CRDN) – Île-de-France in brief …
1/3 245 positive tests for congenital hypothyroidism*
1/15 338 positive tests for congenital adrenal hyperplasia*
1/9 925 positive tests for cystic fibrosis*
1/5 624 positive tests for phenylketonuria (and moderate hyperphenylalaninemia)*
1/411 positive tests for major sickle cell syndromes*
* data valid for 2021